Project - Neuropathology and clinicopathological correlations in FTD and related disorders, and preclinical models of dementia
List all Chief investigators and associate investigators
Lars Ittner, Jillian Kril, Glenda Halliday
Research Project Abstract
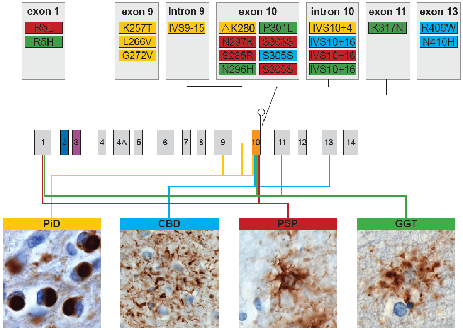
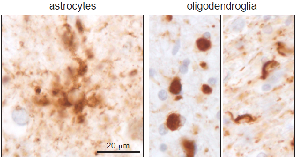
Frontotemporal lobar degeneration (FTLD) is a neurodegenerative disease affecting ~15 per 100,000 people between the ages of 45-64 years. It is characterised by a progressive deterioration in personality and behaviour and/or language skills. While the distribution and severity of neuronal loss in FTLD underlies the clinical symptoms, the contribution of astrocytes and oligodendroglia, which play vital roles in maintaining neural function, are under appreciated. Unlike most other neurodegenerative diseases, glia are involved in the primary pathology of FTLD. Both cell types abnormally aggregate phosphorylated tau and the distribution, morphology, cellular compartment affected and the biochemical composition of inclusions differ substantially to produce distinct pathological subtypes of FTLD-tau. These differences suggest a pivotal role for altered glial function in the pathogenesis of FTLD-tau. The overarching aim of this study is to explore the pathological heterogeneity of astrocytes and oligodendroglia in FTLD. The type and severity of pathology will be examined in a pathological FTLD cohort and preclinical models. This study will provide important insights into the molecular understanding of the distinctive FTLD-tau subtypes and the contribution of astrocytes and oligodendroglia in the pathogenesis of FTLD.
Project with a disease tag
Dementia, MND, FTD, Ageing
Challenges within the field
Predicting the underlying pathology and cell type affected responsible for disease during life is challenging.
Despite the critical roles of astroglia and oligodendroglia in maintaining neural function, they have not been conceptualized in clinical, neuroimaging or biomarker studies for neurodegenerative diseases with pathological tau deposition.
Research Project Description
Shelley’s research focuses on the neuropathology and disease mechanisms underlying frontotemporal dementia, ageing and a range of neurodegenerative disorders. This work involves the investigation of protein abnormalities and cell types affected to determine the selective regional and cellular vulnerability in these disorders, and associated clinicopathological correlations. To address these questions, Shelley uses a combination of histology, immunohistochemistry and quantitative neuropathology in well-characterised human brain tissue samples and preclinical models of dementia.
Research Objectives
To determine the selective regional and cellular vulnerability and associated clinicopathological correlations in FTD and related disorders and preclinical models of these disorders.
Key Publications from this project
- Forrest SL, Kril JJ, Wagner S, Hönigschnabl S, Reiner A, Fischer P, Kovacs GG. (2019). Chronic traumatic encephalopathy (CTE) is absent from a European community-based ageing cohort while cortical ageing-related tau astrogliopathy (ARTAG) is highly prevalent. J Neuropathol & Exp Neurol 78(5):398-405; doi: 10.1093/jnen/nlz017.
- Forrest SL, Kril JJ, Halliday GM. (2019). Cellular and regional vulnerabilities in frontotemporal tauopathies. Acta Neuropathol (invited review) 138(5):705-727; doi: 10.1007/s00401-019-02035-7.
- Forrest SL, Crockford DR, Sizemova A, McCann H, Shepherd CE, McGeachie AB, Affleck AJ, Carew-Jones F, Bartley L, Kwok JB, Kim WS, Jary E, Tan RH, McGinley CV, Piguet O, Hodges JR, Kril JJ, Halliday GM. (2019). Coexisting Lewy body disease and clinical parkinsonism in frontotemporal lobar degeneration. Neurology 92(21):e2472-e2482; doi: 10.1212/WNL.0000000000007530.
- Burrell JR, Forrest SL, Bak TH, Hodges JR, Halliday GM, Kril JJ. (2016). Expanding the phenotypic associations of globular glial tau subtypes. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring. Alzheimer’s Dement (Amst) 4:6-13.
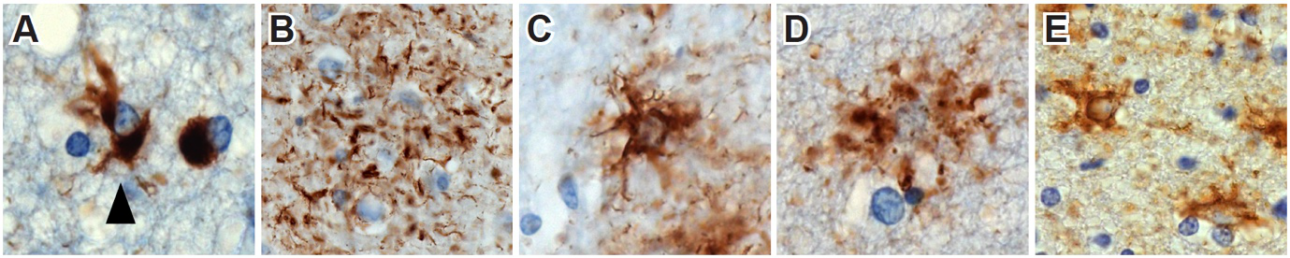
Figure. Astrocytic inclusions in Pick’s disease (A), corticobasal degeneration (B), progressive supranuclear palsy (C), globular glial tauopathy (D) and ageing-related tau astrogliopathy (E).
Key Findings
My publications in this area provide support that the clinical heterogeneity in FTD syndromes is caused by neurodegenerative changes that converge on specific brain regions, giving rise to behavioural, linguistic and motor deficits observed in a variety of syndromes, rather than the type of protein abnormality occurring or the major cell type involved. Furthermore, a number of different molecular and protein pathologies converge on the same neuronal populations and brain regions to produce similar clinical syndromes, which suggests common molecular mechanisms are likely to be involved in neuroanatomical vulnerability.